PROTOCOL 152
Nextflow DSL2
3. Very important to
understand how nextflow works
3.5. Variable declaration in processes
4.3. Old version / previous release
4.4. Installation of DSL1 together with DSL2 already
installed
6. Switch between
different versions of nextflow
6.2. Using Environment Variables
14. Main differences
with Nextflow DSL1
17.3. errorStrategy and maxRetries
19.5. two different file names
21.2. Basic instruction for execution
23. Protection of the
initial files if modified in processes
24.2. Concatenation post parallelized process
24.3. several directories into a channel
24.4. Get the name of a file or
folder together with a path
1. Documentations
https://www.nextflow.io/docs/latest/dsl2.html
https://carpentries-incubator.github.io/workflows-nextflow/
http://www.ens-lyon.fr/LBMC/intranet/services-communs/pole-bioinformatique/bioinfoclub_list/nextflow-dsl2-laurent-modolo
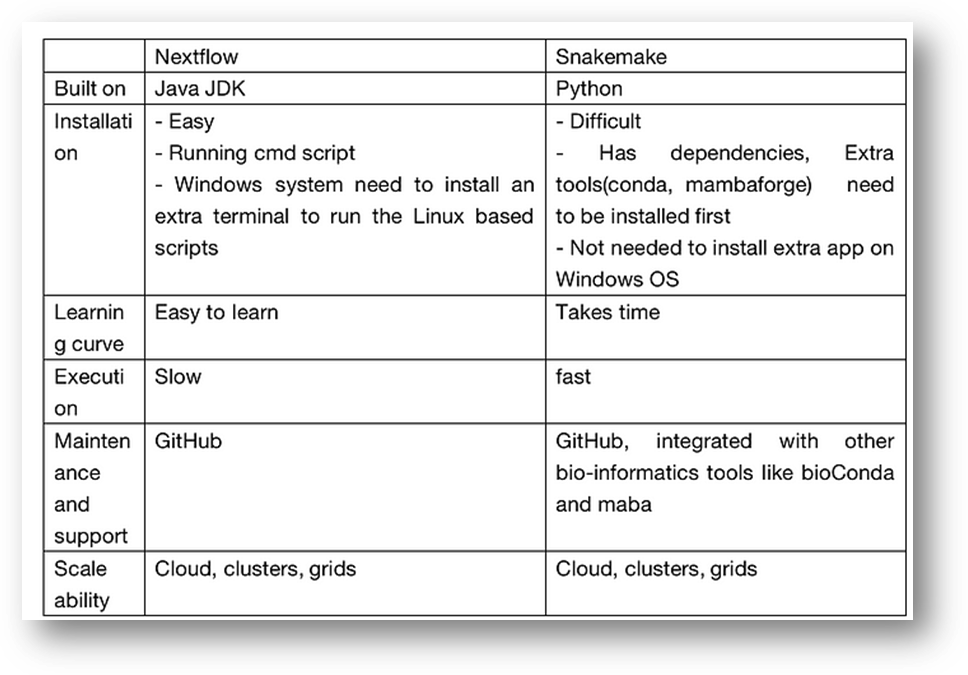
Pro
and con of Nextflow, comparison with snakemake:
2. Acronyms
DAG Directed
Acyclic Graph
DSL Domain Specific Language. See https://en.wikipedia.org/wiki/Domain-specific_language
DSL2 Version 2 of Nextflow. See https://www.nextflow.io/blog/2020/dsl2-is-here.html
nf nextflow
3. Very important to understand how nextflow works
3.1. Names
of the files
The
nextflow pipeline file is named by default main.nf.
The
nextflow config file must are named by default nextflow.config.
It is possible to modify these two names. But it will not work if the idea is to work from a distant repository (see section Error! Reference source not found.).
3.2. work
folders
Nextflow create a folder for each process of the .nf file (or each parallelization of each process) in the work directory. For instance, 00/5bf63033e13094f180fe69c583ce3c. Then, everything of the process is performed in this folder, which becomes the working environment (a kind of home). If INPUT is present, then files or values are imported. For the files, a symbolic link is created pointing to the initial file (in another folder of the work directory, or in a dataset folder). All the new files are created here. If publishDir is used, then the files referenced in OUTPUT are copy/paste into the designed publishDir folder.
3.3. symbolic
links
Warning, because of the symbolic links, every imported file that is modified by the process is not only modified in the work folder but also were the link point, meaning that initial input data can be modifed !
To prevent that:
1) always use input files that are "write" protected (read and execute only).
2) Copy the input file in the process before working:
input:
file fs
script:
"""
cp ${fs} tempo.txt
chmod 777 tempo.txt
...
See also section Error! Reference source not found. for a better example of copy.
3.4. The
resume option
A strong advantage of nextflow is to rerun the pipeline using the -resume option of the run command. In such case, all the processes that are cache 'true' are not run but linked to the previous same process execution, which save lots of time!
However, all the variables of the .config used in the process must be declared in the input section of the process. Otherwise, nextflow will not rerun a cache 'true' process using a variable modified in the .config file.
3.5. Variable
declaration in processes
The scope of a process is such that if a variable is not in the input section, it looks for variables in the working environment of nextflow, which has notably sourced the env{} environment of the config file. Thus, it is not mandatory to use the input section to declare variables. However, in that case, nextflow will not rerun a cache 'true' process using a variable modified in the .config file.
4. Installation
update
using DSL1
The best is to install in our home/bin/ the nextflow software and dependencies (java and singularity).
4.1. Local
Linux kernel
4.1.1. Linux
and WSL2
Java install: see protocol 165.
Nextflow install:
sudo chmod 777 /usr/local
sudo chmod 777 /usr/local/bin
cd /usr/local/bin
sudo curl -s https://get.nextflow.io |
bash
chmod 777 nextflow
Of note, this is specified but Fred Lemoine confirmed that does not work:
wget -qO-
https://github.com/nextflow-io/nextflow/releases/download/v21.04.1/nextflow-21.04.1-all
PATH checking:
echo $PATH # to check that the string
"/usr/local/bin" is included between colons in this variable
PATH update (if required)
export PATH="/usr/local/bin:$PATH"
# add the nextflow path into this variable
Apptainer has to be
installed. See protocol 135.
4.1.2. MacOS
Check Java version:
java -version # Check that java 11 to 18
is installed
Java install:
https://download.oracle.com/java/17/latest/jdk-17_macos-x64_bin.tar.gz
Then open the terminal and check the Java version again.
If several versions are installed, it is possible to select the appropriate version like this:
VERSION=17 #
version to use
/usr/libexec/java_home -V
echo $JAVA_HOME
export JAVA_HOME=$(/usr/libexec/java_home
-v $VERSION)
java -version
Then check if wget is present, otherwise:
sudo port install wget
Then:
wget -qO- https://get.nextflow.io | bash
# Weirdly, nextflow command is installed
in the Users directory. Thus, check that install is correct:
cd /Users/bruhns/
nextflow # should display options. If
not:
echo $PATH
export
PATH="/Users/bruhns:$PATH"
echo $PATH
nextflow # should display options
Apptainer has to be installed. See protocol 135.
4.2. Cluster
Connect to sonic.
Then
cd $ZEUSHOME
cd bin
module load java
curl -s https://get.nextflow.io | bash
Of note, this is specified but Fred Lemoine confirmed that does not work:
wget -qO-
https://github.com/nextflow-io/nextflow/releases/download/v21.04.1/nextflow-21.04.1-all
Then, update the for_sonic.profile and for_sonic.bashrc
export
PATH="/pasteur/zeus/projets/p01/BioIT/gmillot/bin:$PATH" # for
nextflow
alias nextflow="module load java/13.0.2 ; nextflow"
4.3. Old
version / previous release
curl -s https://get.nextflow.io | bash -s
-- -v 20.10.0 # replace 20.10.0 by the desired version
4.4. Installation of DSL1 together with DSL2 already installed
Of note, java 11 works for both DSL1 and DSL2, but when using DSL2, is is better to use the last compatible java version for security reasons.
4.4.1. classic
way
When nextflow (DSL2 only is already installed):
wget -qO nextflow
https://github.com/nextflow-io/nextflow/releases/download/v20.10.0/nextflow #
replace 20.10.0 by the desired version
sudo mv nextflow
/usr/local/bin/nextflow-dsl1
nextflow-dsl1 -v
Then to use the old version of nextflow (DSL1):
sdk default java 11.0.22-amzn #see
protocol 165 for java
nextflow-dsl1 run main.nf
To use the nex DSL2 version of nextflow:
sdk default java 11.0.22-amzn #see
protocol 165 for java
nextflow-dsl1 run main.nf
4.4.2. sdk
management
Install SDKMAN if you haven't installed it yet.
curl -s "https://get.sdkman.io"
| bash
source
"$HOME/.sdkman/bin/sdkman-init.sh"
Install the versions of Nextflow you want. For instance, if you want to install Nextflow 20.10.0 and 21.04.0, you would run the following commands:
sdk install nextflow 20.10.0
sdk install nextflow 23.04.4.5881
Set the desired Nextflow version as the default. For instance, to set 20.10.0 as the default, you would run:
sdk default java 11.0.22-amzn #see
protocol 165 for java
sdk default nextflow 20.10.0 # or sdk use
nextflow 20.10.0
sdk default java 11.0.22-amzn #see
protocol 165 for java
sdk default nextflow 20.10.0 # or sdk use
nextflow 20.10.0
5. Upgrade
nextflow -self-update
Update the version where it is already installed.
6. Switch between different versions of nextflow
6.1. Using
path
/path/to/nextflow-<version>/nextflow
run your_workflow.nf
Replace /path/to/nextflow-<version>/ with the path where the specific version of Nextflow is installed, and your_workflow.nf with the actual filename of your Nextflow workflow.
6.2. Using
Environment Variables
You can also use the NXF_VER environment variable to set the Nextflow version for a particular session. For example:
export NXF_VER=20.10.0
nextflow run your_workflow.nf
This will set the Nextflow version to 20.10.0 for the current terminal session.
7. DSL1 or DSL2 by default
As of version 22.03.0-edge Nextflow defaults to DSL 2 if no version is specified explicitly. You can restore the previous behavior setting in into your environment the following variable:
export NXF_DEFAULT_DSL=1
See section 8.1.
8. nexflow
usage
8.1. DSL1
and DSL2
Always start a .nf script by:
nextflow.enable.dsl=2
With value 1 or 2 depending on the desired DSL.
8.2. Commands
clean Clean up project cache and work directories
clone Clone a project into a folder
config Print a project configuration
console Launch Nextflow interactive console
drop Delete the local copy of a project
help Print the usage help for a command
info Print project and system runtime information
kuberun Execute a workflow in a Kubernetes cluster (experimental)
list List all downloaded projects
log Print executions log and runtime info
pull Download or update a project
run Execute a pipeline project
self-update Update nextflow runtime to the latest available version
view View project script file(s)
Example:
nextflow run main.nf
8.3. Main
options
-C Use the specified configuration file(s) overriding any defaults
-D Set JVM properties
-bg Execute nextflow in background
-c, -config Add the specified file to configuration set
-d, -dockerize Launch nextflow via Docker (experimental)
-h Print this help
-log Set nextflow log file path
-q, -quiet Do not print information messages
-syslog Send logs to syslog server (eg. localhost:514)
-v, -version Print the program version
Example:
nextflow -v
8.4. Main
run options
-cache Enable/disable processes caching
-h, -help Print the command usage Default: false
-main-script The script file to be executed when launching a project directory or repository
-name Assign a mnemonic name to the run pipeline
-resume Execute the script using the cached results, useful to continue executions that were stopped by an error. Warning: as soon as a cache = false in a process, all the process that uses a channel from this process will be re-executed. Thus, if the first process is cache = false and that it channels toward the second process, then every process would be re-executed
-r, -revision Revision of the project to run (either a git branch, tag or commit SHA number)
-test Test a script function with the name specified
-N, -with-notification Send a notification email on workflow completion to the specified recipients
-with-weblog Send workflow status messages via HTTP to target URL
-w, -work-dir Directory where intermediate result files are stored
Example:
nextflow run main.nf -resume
9. Basic
description
See https://www.nextflow.io/docs/latest/process.html?highlight=tuple

Nextflow is a DSL (see section 2).
Comparison with other workflow managers:
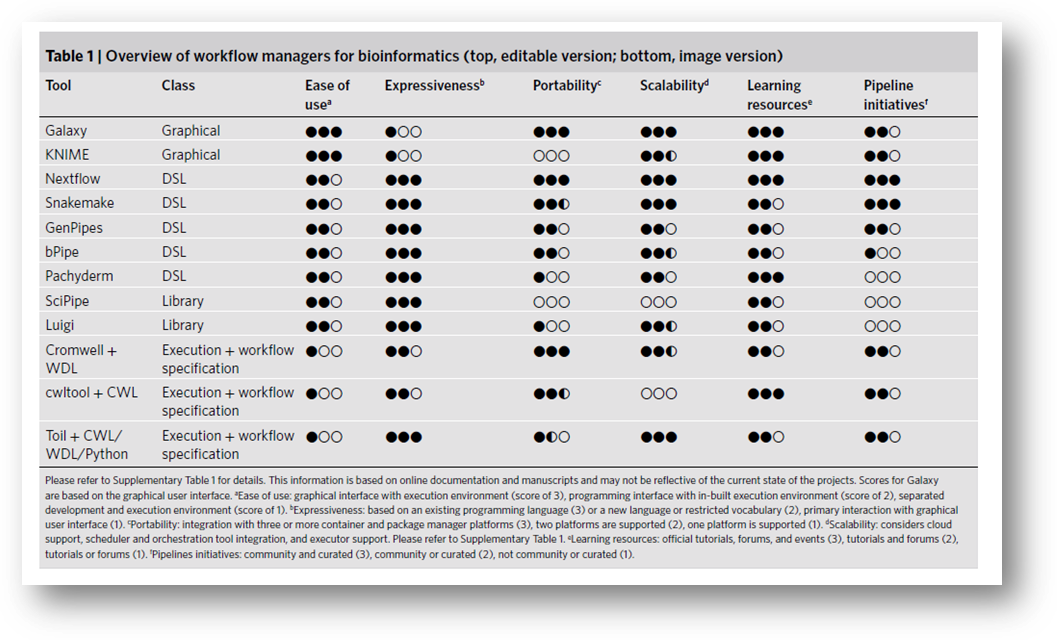
https://www.nature.com/articles/s41592-021-01254-9
Nextflow uses the Groovy language.
https://www.nextflow.io/docs/latest/script.html#language-basics
https://groovy-lang.org/syntax.html
https://www.tutorialspoint.com/groovy/index.htm
Regex:
https://www.nextflow.io/docs/latest/script.html#regular-expressions
https://web.archive.org/web/20170621185113/http://naleid.com/blog/2008/05/19/dont-fear-the-regexp/
https://docs.oracle.com/javase/tutorial/essential/io/fileOps.html#glob
https://docs.oracle.com/javase/7/docs/api/java/util/regex/Pattern.html
10. Variables
10.1. Empty
file
This can be convenient when a file is missing:
tbi_file = file("NULL")
11. Console
Very useful to test groovy code.
On the cluster:
module load java
nextflow console
Nice Web console:
https://groovyide.com/playground
http://groovyconsole.appspot.com/edit/34004
12. Main
groovy commands
12.1. basic
x.first() If parallelization due to another channel in the same input section, specify that the x channel has to be used each time
x.name take the name of the variable x. To assign the name, use val(x.name) into y
${x} value of the variable x, defined at the beginning of the file
// comment
channel. function that defines a channel. Must be followed by another function (see below)
channel.from(1,2) channel for the value 1 and 2
Channel.fromPath("x") define
a channel from the path x. Warning: a channel can be called only once, contrary
to variables
Channel.fromFilePairs("x") take
as argument a pathway of two files. If several couples of files, regular
expressions are used. The result is a tuple
file() define a path variable. For instance file("x") defines the path x as a variable, that can be used several times in a nextflow script if defined at the beginning of a nf script
x.first() take
the first element of channel x
x.collectFile() collect
all the files of channel x, means that it waits for the end of the
parallelization of the process using the output channel x. Warning: see section
Error! Reference source not
found.. Optional: x.collectFile(name:"all_consensus.fasta",
skip: 1, keepHeader: true)
x.collect()
x.mix(y, z)
x.flatten() split
a list of file inside a single channel x into several channels with one file
per channel
publishDir "x/", mode: 'copy' copy the output files defined in the output section into the x folder. The mode: 'copy' precises that we do not want a symbolic file into the x folder pointing to the work folder (default) but that we want a real copy. This avoid breaking symbolic links if the wok folder is removed for instance
tuple create a channel containing
several info (like a list). Example: tuple val(name), file(f1), file(f2) from data
val(y) from x recover a value from x and assign to y. Can be an integer, character,
etc.
file(y) from x recover a file from x and assign to y. In input: section
12.2. condition
|| ou
&& and
!= different
== equal
if(){}else if(){}else{} if
13. Condition
with if(){}
In the workflow{} section, it is possible to check variables and so on:
workflow {
if( ! (sample_path in String) ){
error "\n\n========\n\nERROR IN NEXTFLOW EXECUTION\n\nINVALID
sample_path PARAMETER IN repertoire_profiler.config FILE:\n${sample_path}\nMUST
BE A SINGLE CHARACTER STRING\n\n========\n\n"
}else if( ! (file(sample_path).exists()) ){
error "\n\n========\n\nERROR IN NEXTFLOW EXECUTION\n\nINVALID
sample_path PARAMETER IN repertoire_profiler.config FILE (DOES NOT EXIST):
${sample_path}\nIF POINTING TO A DISTANT SERVER, CHECK THAT IT IS MOUNTED\n\n========\n\n"
}
}
Warning: in the workflow{} section, if(){} are checked immediately, wherever the code is written. This means that end of processes are not waited. For instance:
process igblast{
input:
path f
output:
path "*.out"
script:
"""
$f > ${f.baseName}.out
"""
}
igblast.out.collectFile().view()
workflow{
fs_ch = Channel.fromPath("/Users/Gael/f*.test", checkIfExists:
false) // two files made of different nb of lines
igblast(fs_ch)
if( igblast.out.collectFile().countLines() != fs_ch.collectFile().countLines()
){
error "\n\n========\n\nERROR IN NEXTFLOW EXECUTION\n\nTHE NUMBER OF
LINES IN THE OUTPUT OF IGBLAST IS NOT EQUAL TO THE NUMBER OF LINES IN THE
SUBMITTED FILES\n========\n\n"
}
}
The
error message will come even before the igblast.out.collectFile().view().
The
only way to have the condition testing waiting for the end of a process is to
use subscribe(). Example:
process igblast{
input:
path f
output:
path "*.out"
script:
"""
$f > ${f.baseName}.out
"""
}
igblast.out.collectFile().view()
workflow{
fs_ch = Channel.fromPath("/Users/Gael/f*.test", checkIfExists:
false) // two files made of different nb of lines
igblast(fs_ch)
nblines1 = igblast.out.collectFile().countLines()
nblines2 = fs_ch.collectFile().countLines()
nblines1.combine(nblines2).subscribe{n1,n2 -> if(n1!=n2){
error "\n\n========\n\nERROR IN NEXTFLOW EXECUTION\n\nTHE NUMBER OF
LINES IN THE OUTPUT OF IGBLAST IS NOT EQUAL TO THE NUMBER OF LINES IN THE
SUBMITTED FILES\n========\n\n"
}
}
14. Main differences with Nextflow DSL1
See protocol 136 for nextflow DSL1.
1) Now custom functions can be designed in the .nf script. See https://www.nextflow.io/docs/latest/dsl2.html#function
2) Process section identical except that the from and into channel declarations have to be omitted.
3) Output section. See section 19.
4) Processes are executed using a new section workflow{}. See https://www.nextflow.io/docs/latest/dsl2.html#workflow
3.1) Workflow inputs are always channels by definition. If a basic data type is provided instead, such as a number, string, list, etc, it is implicitly converted to a value channel.
3.2) A workflow definition which does not declare any name (also known as implicit workflow) is the entry point of execution for the workflow application.
3.3) Several workflow sections can be defined and then used inside another workflow section.
3.4) Channels can be used several times (no need to define same channels with different names).
5) Modules. See https://www.nextflow.io/docs/latest/dsl2.html#modules
6) Other things. See https://www.nextflow.io/docs/latest/dsl2.html#dsl2-migration-notes
7) Deprecation:
1) from or file() use path or path() instead (notably in input and output processes).
8) Values or path specified in the env{} section of the .config file do not have to be declared as input in processes, and as arguments of processes in the workflow{} section. Example in the .config file (on value and one path parameter):
env {
REPLACE_SPACE_CONF="_._"
BASH_FUNCTIONS_CONF="/mnt/c/Users/Gael/Documents/Git_projects/little_bash_functions/little_bash_functions.sh"
}
And in the .nf file (no input declared):
process lodscore {
label 'bash'
script:
"""
eval lodscore.sh
"""
}
workflow {
lodscore(
)
}
However, is something is declared in the input section of a process, it must also be declared as argument in the core of the process in the workflow{} section:
process lodscore {
label 'bash'
input:
path BASH_FUNCTIONS_CONF
script:
"""
eval lodscore.sh
"""
}
workflow {
lodscore(
BASH_FUNCTIONS_CONF
)
}
9) for parallelization of a process for each file in a folder, one way is to write this:
process lodscore {
label 'bash'
input:
path DIR_ch
script:
"""
#!/bin/bash -ue
eval lodscore.sh
"""
}
workflow {
if( ! file(DIR).exists()){
error "\n\n========\n\nERROR IN NEXTFLOW EXECUTION\n\nINVALID DIR
PARAMETER IN linkage.config FILE (DOES NOT EXIST): ${DIR}\nIF POINTING TO A
DISTANT SERVER, CHECK THAT IT IS MOUNTED\n\n========\n\n"
}else{
DIR_ch = Channel.fromPath("${DIR}/*.*", checkIfExists: false)
}
lodscore(
DIR_ch
)
}
10)
Gael
MILLOT la prochaine version de nextflow int grera une commande apptainer
utiliser la place de singularity. Par contre, elle ne g rera plus DSL1.
Si tu
veux d j tester, tu peux faire:
export NXF_EDGE=1
nextflow self-update
15. Functions
and operators
Functions are objects that can modify, read, etc. an object. Example of function usage using the x object:
x = "Hello"
print(x)
Most functions are from the groovy language.
Custom (home made) functions:
https://www.nextflow.io/docs/latest/dsl2.html#function
Operators allow to manipulate channels (see https://www.nextflow.io/docs/latest/operator.html). Example of operator usage using the x channel:
x = Channel.fromPath("./file.txt")
x.view()
Thus, operators are used with a dot, but not groovy or home made functions.
16. docker
images
Nextflow uses containers to run processes. Example on how a docker image is set in the nextflow.config file:
process {
withLabel: bash {
container='gmillot/alohomora_merlin_v2.1:gitlab_v9.5'
cpus=1
memory='6G'
}
}
1) Nextflow can automately convert docker images to singularity, using this in the nextflow.config file
singularity {
enabled = true
autoMounts = true // automatically mounts host paths in the executed
container
if(system_exec == 'slurm' || system_exec == 'slurm_local'){
runOptions = '--no-home --bind /pasteur -B /run/shm:/run/shm'
}else{
runOptions = '--no-home -B /run/shm:/run/shm' // --no-home prevent
singularity to mount the $HOME path and thus forces singularity to work with
only what is inside the container
}
//runOptions = '--home $HOME:/home/$USER --bind /pasteur' // provide any
extra command line options supported by the singularity exec. Here,
fait un bind de tout /pasteur dans /pasteur du container. Sinon pas d acc s
if(system_exec == 'slurm'){
cacheDir = '/pasteur/zeus/projets/p01/BioIT/gmillot/singularity' // name
of the directory where remote Singularity images are stored. When rerun, the
exec directly uses these without redownloading them. When using a computing
cluster it must be a shared folder accessible to all computing nodes
}else if(system_exec == 'slurm_local'){
cacheDir = 'singularity' // "$baseDir/singularity" can be used
but do not forget double quotes.
}else{
cacheDir = '/mnt/c/Users/gmillot/singularity' //
"$baseDir/singularity" can be used but do not forget double quotes.
}
}
2) Warning: nextflow does not use the entrypoint of the images. It systematically uses --entrypoint bash, i.e., open the container in the system environment.
17. Process
17.1. executor
Different way to execute:
Local (default), SGE, slurm, PBS, Kubernetes, AWS Batch, Google, etc.
process igblast {
executor ="local"
17.2. container
Warning: by default, execute on the local environment (no container). For instance, using executor ="slurm", the process is run on the head of the cluster. If modules are imported in my session before using nextflow run, they are found. But this is not a proper way to work.
process igblast {
container = "evolbioinfo / goalign : v0 .3.2"
17.3. errorStrategy
and maxRetries
process igblast {
maxRetries=3
errorStrategy = {task.exitStatus in 137..143 ? 'retry' : 'terminate' }
Here is to indicate that if the exit status is between 137 and 143, then retry the number of times indicated by maxRetries. Otherwise terminate.
18. input
18.1. Example
Warning: inputs defined in a process inside workflow{} must be in the same order as in the input section of the process before workflow{}. Example:
process igblast {
...
input:
val("CHROMO_NB"), path("DIR") // from channel x1_ch
val x2
...
}
workflow {
igblast(
x1_ch,
x2
)
}
Here, the channel x1_ch is a tuple that is split in the input section of the igblast process.
18.2. Tuple
process fisher {
input:
tuple val(region2), path(vcf) // parallelization expected for each value
of region_ch
workflow {
vcf_ch = Channel.fromPath(sample_path) # sample_path from the
nextflow.config file
if(region == 'none'){ # region from the nextflow.config file
region_ch = Channel.from("chr1", "chr2",
"chr3", "chr4", "chr5", "chr6",
"chr7", "chr8", "chr9", "chr10", "chr11",
"chr12", "chr13", "chr14", "chr15",
"chr16", "chr17", "chr18", "chr19",
"chr20", "chr21", "chr22", "chr23",
"chr24", "chr25", "chrY", "chrX",
"chrM")
}else{
if(region =~ /,/){
tempo = region.replaceAll(':.+,',
',')
}else{
tempo = region
}
tempo2 = tempo.replaceAll(':.+$', '')
tempo3 = tempo2.replaceAll(' ', '')
tempo4 = tempo3.split(",") // .split(",")
split according to comma and create an array
https://www.tutorialspoint.com/groovy/groovy_split.htm
region_ch = Channel.from(tempo4)
}
fisher(
region_ch.combine(vcf_ch2) // combine the vcf file and each value of
region_ch
)
}
19. output
19.1. Quotes
and no quotes
Warning: in the output section, variable with no quotes means that the variable is a nextflow variable. Quotes mean that the output is a string and that glob (wildcard) can be used. Strings work for path outputs. However, it is not possible to add a variable created in the script section as val output. Only stdout can do it but forward all the stdout of the script section.
Example:
process igblast {
...
output:
val TEMPO
path "*.log"
script:
TEMPO=4
TEMPO without quotes produces an error because TEMPO is not defined as a nextflow variable, and TEMPO with quotes send "TEMPO".
See https://www.nextflow.io/docs/latest/process.html?highlight=tuple#output-type-tuple
See section 19.2 to solve this.
19.2. Stdout
in output
The only way to have a value in output, potentially sent into a channel is to use the stdout of the script section. Example:
process standardout_test {
label 'bash' // see the withLabel: bash in the nextflow config file cache 'true'
input:
path vcf_header // no parall
output:
stdout
script:
"""
#!/bin/bash -ue
cat ${vcf_header} | wc -l
"""
}
workflow{
standardout_test(
header.out.vcf_header_ch
)
standardout_test.out[0].view()
}
The number of lines of the vcf header is the stdout and is emitted in vcf_header_line_nb
Warning: \n (end of lines) can impair the correct use of the variable. See trim in groovy or printf in bash to solve that.
19.3. Emit in
channel
If output are not named (emitted), then:
process igblast {
...
output:
path "*.tsv"
path "*.log"
Then, outputs of the igblast process can be used like this:
igblast.out[0].view() // for the first
output of igblast
igblast.out[1].view() // for the second
output of igblast
If output are named, then:
process igblast {
...
output:
path "*.tsv", emit: tsv_ch1
path "*.log", emit: log_ch
Then, outputs of the igblast process can be used like this:
igblast.out.tsv_ch1.view()
igblast.out.log_ch.view()
Thus, channels in outputs are not necessarily named and emit only facilitate the use of a channel several times.
19.4. Optional
process igblast {
...
output:
path "*.tsv", emit: tsv_ch1, optional: true
With or without emit.
19.5. two
different file names
Use this writting if the output file is either file.tsv or NULL:
path '{file.tsv,NULL}', includeInputs:
true, emit: meta_file_ch
includeInputs: true means that any files with the glob of {file.tsv,NULL} will be added into the channel.
20. Workflow
writing
2 options:
process writeFiles {
input :
val odd
output :
file "*. txt "
script :
"""
echo " Value = ${ odd }" > file_$
{ odd }. txt
"""
}
workflow {
ch = Channel .of( 1, 3, 5, 7 )
oddfiles = writeFiles(ch) // equivalent to
}
workflow {
ch = Channel .of( 1, 3, 5,
7 )
writeFiles(ch)
oddfiles =
writeFiles.out[0]
}
oddfiles gather all the outputs, which can be called using [0], [1], etc.
oddfiles is a channel.
Warning: emit is optional. It is just to give a name to the channel.
process writeFiles {
input :
val odd
output :
file "*. txt ", emit: out_ch
script :
"""
echo " Value = ${ odd }" >
file_$ { odd }. txt
"""
}
workflow {
ch = Channel .of( 1, 3, 5, 7 )
writeFiles(ch)
oddfiles = writeFiles.out.out_ch
}
21. running
21.1. Defaults
names
Nextflow recognizes main.nf and nextflow.config as the two nf and config files by default.
When using nextflow run, this means that:
1) it is not mandatory to specify the config file name if it is in the same path as the main.nf file and if it is named nextflow.config. Example:
nextflow run /home/gmillot/tmp/main.nf
2) Running from a distant repository, the two files are recognized
nextflow run
https://gitlab.pasteur.fr/gmillot/14985_loot -r master
21.2. Basic
instruction for execution
21.2.1.
Local running
Where the instruction is executed is important. Indeed, several files and folders are created at this place: result, work, .nexflow, etc. Once the location chosen using cd, do:
nextflow run /home/gmillot/tmp/main.nf
The main.nf file is executed. Parameters are in the nextflow.config file in the same folder as main.nf.
To specifiy the file name and path of the config file:
nextflow run /home/gmillot/tmp/main.nf -c
/home/gmillot/tmp/nextflow.config
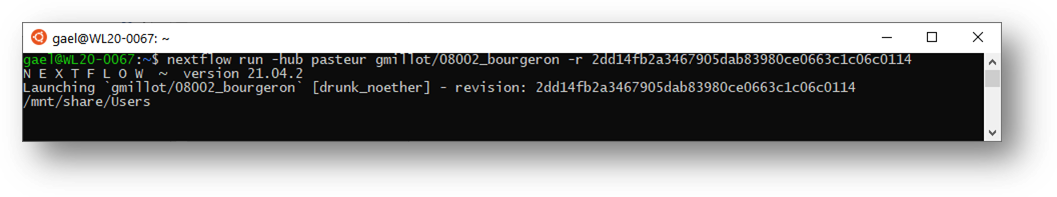
See section 21.2.4 if nothing happens, like:
21.2.2.
Using a distant repository
If the linkage.nf file in a public gitlab repository
Run the following command from where you want the results:
nextflow run -hub pasteur
gmillot/genetic_linkage -r v1.0.0
-r for the tagged version of the file.
Warning: the .config file used by default is the one present in the distant repository. The config file can be on the local computer but -c option has to be used.
If the linkage.nf file in a private gitlab repository
Create the scm file:
providers {
pasteur {
server = 'https://gitlab.pasteur.fr'
platform = 'gitlab'
}
}
And save it as 'scm' in the .nextflow folder. For instance in:
\\wsl$\Ubuntu-20.04\home\gmillot\.nextflow
Warning: ssh key must be set for gitlab, to be able to use this procedure.
Then run the following command (example: from here \\wsl$\Ubuntu-20.04\home\gmillot):
nextflow run -hub pasteur
gmillot/genetic_linkage -r v1.0.0
21.2.3.
On clusters
Warning: the workflow{} section is made of processes that are executed in nodes but the inter process code is executed on the node where the nexflow run command is executed. This means that the head od the cluster can be saturated if used for nexflow run.
21.2.4.
Check that a distant server
is mounted
See protocol 140.
Example:
DRIVE="C"
sudo mkdir /mnt/share
sudo mount -t drvfs $DRIVE: /mnt/share
Warning: if no mounting, it is possible that nextflow does nothing, or displays a message like:
Launching
`linkage.nf` [loving_morse] - revision: d5aabe528b
/mnt/share/Users
21.2.5.
Error messages and solutions
1)
Unknown
error accessing project `gmillot/genetic_linkage` -- Repository may be
corrupted:
/pasteur/sonic/homes/gmillot/.nextflow/assets/gmillot/genetic_linkage
Purge using:
rm -rf
/pasteur/sonic/homes/gmillot/.nextflow/assets/gmillot*
2)
WARN:
Cannot read project manifest -- Cause: Remote resource not found:
https://gitlab.pasteur.fr/api/v4/projects/gmillot%2Flinkage
Make the distant repo public.
In settings/General/Visibility, project features, permissions, check that every item is "on" with "Everyone With Access" and then save the changes.
3)
permission
denied
Use chmod to change the user rights.
4)
Warning: if no mounting, it is possible that nextflow does nothing, or displays a message like:
Launching
`linkage.nf` [loving_morse] - revision: d5aabe528b
/mnt/share/Users
22. -resume
option
The -resume option of nextflow has a considerable benefit, as it prevent the running of long processes. For instance, when changing a parameter in the .config file, nextflow will rerun only the processes that is impacted by the change and all the subsequent ones.
However, this benefit comes with tricky behaviors that must be anticipated:
1) modification inside a file (see section 23).
2) if the code inside the script section of a process A is unchanged and the input are the same, nextflow uses the last work folder created during the last time that the process A has been run. And Nextflow indicates that process A is "cached". This is the expected behavior. However, if modifications of the script section of a process A, goes back to a previous one (word to word), it seems that Nextflow uses the last work folder created using this previous script. It should be logical. But because of 1), this can generate behaviors difficult to understand.
Of note, both the work and .nextflow folders are used by the -resume option. Erasing the work folder is sometimes not sufficient to prevent this option to run. Only the .nextflow deletion will trigger the message:
WARN: It appears you have never run this project before -- Option `-resume` is ignored
23. Protection of the initial files if modified in processes
If an input file is modified in a process, we could imagine that only the file present in the work directory is modified. But due to the symbolic link, the initial file will be also modified. To prevent this:
- NEVER modify a file using the same name. always change the mane of the modified file.
- ALWAYS copy these files in the process that modifies the file, not in another process (to optimize the -resume option of nextflow run).
24. Manipulation
of channels
24.1. Parallelization
The output channel of a parallelized process A, containing a single value or path, will induce parallelization of the next process B if the channel is not assembled between the A and B process in the workflow{}.
24.2. Concatenation
post parallelized process
process igblast {
...
output:
path "*.tsv", emit: tsv_ch1, optional: true
...
}
workflow {
igblast()
igblast.out.tsv_ch1.collectFile()
}
24.3. several
directories into a channel
See the linkage.nf script.
process splitting{
...
output:
path "Group*", emit: group_dir_ch
...
}
workflow {
alohomora(
splitting.out.group_dir_ch.flatten()
}
The flatten() operator splits the list into several objects.
24.4. Get the name of a file or folder together with a path
See the linkage.nf script.
24.4.1.
Simplest instruction
If the input is several path, generated by the previous process like this:
output:
path "Group*", emit: group_dir_ch
Then the name can be recover just by using the file method baseName: in the output and in the script section:
process alohomora {
...
input:
path group_dir_ch
output:
tuple val(group_dir_ch.baseName), path("Group*/merlin/c*"),
emit: chr_dir_ch // Warning: a tuple with a single group_name associated with
23 path
script:
"""
#!/bin/bash -ue
GROUP_NAME=${group_dir_ch.baseName}
}
workflow {
alohomora(
splitting.out.group_dir_ch.flatten()
)
}
24.4.2.
Pity but map{} does not work
process splitting{
...
output:
path "Group*", emit: group_dir_ch
...
}
process alohomora{
...
input:
tuple val(group_name), path(group_dir_ch)
...
}
workflow {
alohomora(
splitting.out.group_dir_ch.flatten().map{it -> [it.baseName(), it]}
}
The map{} operator convert the path into a tuple according to what is written between brackets.
process alohomora {
...
input:
tuple val(group_name), path(group_dir_ch)
output:
tuple val(group_dir_ch.baseName), path("Group*/merlin/c*"),
emit: chr_dir_ch
The output tuple can also be:
tuple val(group_name), path("Group*/merlin/c*"), emit:
chr_dir_ch
Here the channel is complicate because it is intricated lists, like:
Channel.of(
['a', ['p', 'q'], ['u','v']],
['b', ['s', 't'], ['x','y']]
)
To correctly split the channel during the next step, the operator to use is transpose():
[a, p, u]
[a, q, v]
[b, s, x]
[b, t, y]
So the instruction in the workflow{} would be:
alohomora.out.chr_dir_ch.transpose()
No need to flatten if we use a tuple as input.
25. Simple
writting
26. config
file
Can be called by nextflow when in the same repertoire as main.nf or if the full path is specified with the -c option:
nextflow run -c /mnt/c/Users/Gael/Documents/Git_projects/repertoire_profiler/nextflow.config
main.nf
Example of nextflow.config file:
/*
#########################################################################
##
##
##
repertoire_profiler.config ##
##
##
##
Gael A. Millot
##
##
Bioinformatics and Biostatistics Hub ##
##
Computational Biology Department ##
##
Institut Pasteur Paris ##
##
##
#########################################################################
*/
/*
#########################################################################
##
##
##
Parameters that must be set by the user ##
##
##
#########################################################################
*/
/*
##########################
## ##
##
Ig Clustering ##
## ##
##########################
*/
env {
sample_path =
"/mnt/c/Users/Gael/Documents/Git_projects/repertoire_profiler/dataset/ig_clustering_test_1/VH"
// single character string of the path of the fasta files directory. The last /
can be added or not, as it is removed by nextflow file(). Warning: the fasta
names must not start by a digit, before alakazam::readChangeoDb() function is
fixed by the maintainer. Example :
sample_path="/mnt/c/Users/Gael/Documents/Git_projects/repertoire_profiler/dataset/A1_IgG_H_fw.fasta"
or sample_path="/pasteur/appa/homes/gmillot/dataset/20210707_AV07016_HAD-III-89_plate3_IgK_sanger_seq".
Example with spaces in the path: sample_path="/mnt/x/ROCURONIUM PROJECT/01
Primary data/04.Repertoire analysis/SORT1/SORT1
Seq-original/xlsx_to_fasta_1669018924/All/VL"
igblast_database_path = "germlines/imgt/mouse/vdj" // single
character string of the path of the database provided by igblast indicating a
folder of fasta files, WITHOUT the last /. See \\wsl$\Ubuntu-20.04\home\gael\share
for the different possibilities of paths. Example:
igblast_database_path="germlines/imgt/human/vdj". Warning: change
this code in the .nf file " MakeDb.py igblast -i \${FILE}_igblast.fmt7 -s
${fs} -r \${REPO_PATH}/imgt_human_IGHV.fasta
\${REPO_PATH}/imgt_human_IGHD.fasta \${REPO_PATH}/imgt_human_IGHJ.fasta
--extended" if the present path is modified
igblast_organism = "mouse" // single character string of the
value of the --organism option of AssignGenes.py igblast. Example:
igblast_organism="human". Example: igblast_organism="mouse"
igblast_loci = "ig" // single character string of the value of
the --loci option of AssignGenes.py igblast. Example:
igblast_loci="ig"
igblast_aa = "false" // single character string either of the
igblast protein (true) or nucleic (false) aligmnent. Either "true" or
"false" in lower capitals. Warning: "true" means that the
fasta sequences in sample_path must be aa sequences. Example: igblast_aa =
"false". igblast_aa = "true" does not work for the moment
because no j data in the imgt database (compare AssignGenes.py igblast --help and
AssignGenes.py igblast-aa --help). In addition, no junction data are returned
by igblast-aa, which block the clone_assignment process
igblast_files = "imgt_mouse_IGHV.fasta imgt_mouse_IGHD.fasta
imgt_mouse_IGHJ.fasta" // single character string of the files of
igblast_database_path parameter to use (-r option of MakeDb.py igblast). Each
file msut be separated by a single space. Example for human IGH:
igblast_files="imgt_human_IGHV.fasta imgt_human_IGHD.fasta
imgt_human_IGHJ.fasta". Example for human IGK:
igblast_files="imgt_human_IGKV.fasta imgt_human_IGKJ.fasta". Example
for mouse IGK: igblast_files="imgt_mouse_IGKV.fasta imgt_mouse_IGKJ.fasta".
Example for human IGK + IGL: igblast_files = "imgt_human_IGLV.fasta
imgt_human_IGLJ.fasta imgt_human_IGKV.fasta imgt_human_IGKJ.fasta"
clone_nb_seq = "3" // single character string of a positive
integer value greater or equal to 3. Minimun number of non-identical sequences
per clonal group for tree plotting. Example: clone_nb_seq = "5".
Cannot be below 3 as dowser::getTrees(clones, build="igphyml") needs
at least three different sequences
clone_model = "ham" // single character string of the model
used to compute the distance between sequences, before clutering them to clonal
groups. Either "ham", "aa", "hh_s1f",
"hh_s5f", "mk_rs1nf", "mk_rs5nf",
"m1n_compat", "hs1f_compat". Warning. Must be
"aa" if aa are used. See
https://shazam.readthedocs.io/en/stable/topics/distToNearest/ and
https://shazam.readthedocs.io/en/stable/vignettes/DistToNearest-Vignette/
clone_normalize = "len" // single character string of the
normalization used to compute the distance between sequences, before clutering
them to clonal groups. Either "len" or "none". See the
links in clone_model
clone_distance = "0.15" // single character string of a
positive proportion value, setting the distance threhold that defines the
appartenance of 2 sequences to a same clonal group or not. See https://shazam.readthedocs.io/en/stable/vignettes/DistToNearest-Vignette/.
Example: clone_distance = "0.15"
}
/*
########################
## ##
##
Graphics ##
## ##
########################
*/
env {
meta_path =
"/mnt/c/Users/Gael/Documents/Git_projects/repertoire_profiler/dataset/ig_clustering_test_1/metadata.tsv"
// single character string of a valid path of a metadata file for adding
additional info to the trees. Write "NULL" if no metadata to add.
WARNING: the metadata .tsv table must include a first column named
"Label" containing sequence names, i.e., the header of some of the
fasta files from sample_path, without the ">" of the header.
Additionnal columns (quanti or quali) can then be added after the fisrt column
to modify the leafs of the tree. For instance: "KD", or Antibody
name. Example: tree_meta_path =
"/mnt/c/Users/Gael/Documents/Git_projects/repertoire_profiler/dataset/metadata.tsv".
Example: tree_meta_path = "NULL"
meta_name_replacement = "Name" // single character string of
the name of the columns of the table indicated in the tree_meta_path parameter.
This column will be used to replace the sequence names (header of the fasta
files) by more appropriate names. Write "NULL" if not required and if
tree_meta_path is not "NULL". Ignored if tree_meta_path =
"NULL". Ignored if tree_meta_path = "NULL". Example:
tree_meta_name_replacement = "KD". Of note, tree_leaf_size parameter
is ignored if tree_meta_name_replacement is a numeric column ,and
tree_leaf_shape is ignored if the column is another mode
meta_legend = "KD" // single character string of the name of
the columns of the table indicated in the tree_meta_path parameter. This column
will be used to add a legend in trees, in order to visualize an additionnal
parameter like KD, names, etc. Ignored if tree_meta_path = "NULL".
Example: tree_meta_legend = "KD". Of note, tree_leaf_size parameter
is ignored if tree_meta_legend is a numeric column ,and tree_leaf_shape is
ignored if the column is another mode
tree_kind = "rectangular" // single character string of the
kind of tree. Can be "rectangular", "roundrect",
"slanted", "ellipse", "circular", "fan",
"equal_angle", "daylight". See
https://yulab-smu.top/treedata-book/chapter4.html#tree-layouts
tree_duplicate_seq = "FALSE" // single character string
indicating if identical sequences (with difference cell or sequence names) must
be removed from trees or not. Either "TRUE" for keeping or
"FALSE" for removing
tree_leaf_color = "NULL" // single character string of the
color of leaf tip. Ignored if tree_meta_legend parameter is a name of a non
numeric column of the tree_meta_path parameter
tree_leaf_shape = "23" // single character string of the shape
of leaf tip. See http://127.0.0.1:25059/library/graphics/html/points.html
tree_leaf_size = "3" // single character string of the size of
leafs (positive numeric value in mm, the size of the plot being 120 x 120 mm).
Ignored if tree_meta_legend parameter is a name of a numeric column of the
tree_meta_path parameter
tree_label_size = "2" // single character string of the size
of leaf labeling (positive numeric value in mm, the size of the plot being 120
x 120 mm)
tree_label_hjust = "-0.5" // single character string of the
adjustment of leaf labeling (numeric value)
tree_label_rigth = "FALSE" // single character string of the
position of the labeling. Either "TRUE" or "FALSE". Only
works for tree_kind = "rectangular" and users need to use theme() to
adjust tip labels in this case
tree_label_outside = "TRUE" // single character string of the
display of the labeling outside of the plot region (if labels are truncated).
Either "TRUE" or "FALSE"
tree_right_margin = "1.5" // single character string of the
positive numeric value for the right margin in inches, considering 5 inches the
width of the graphic device
tree_legend = "TRUE" // single character string of the display
of the legend. Either "TRUE" or "FALSE"
donut_palette = "NULL" // single character string of the color
palette of the donut. Write "NULL" for default. Example:
donut_palette = "Accent". See https://ggplot2.tidyverse.org/reference/scale_brewer.html#palettes
donut_hole_size = "0.5" // single character string of the
positive proportion of donut central hole, 0 meaning no hole (pie chart) and 1
no plot (donut with a null thickness)
donut_hole_text = "TRUE" // single character string of the
display of the sum of frequencies. Either "TRUE" or "FALSE"
donut_hole_text_size = "14" // single character string of the
positive numeric value of the title font size in mm. Ignored if hole.text is
FALSE
donut_border_color = "gray50" // single character string of
the color of the donut borders. Either "black" or "white".
Example: donut_border_colors = "black"
donut_border_size = "0.2" // single character string of the
size of the donut borders (positive numeric value in mm, the size of the plot
being 120 x 120 mm)
donut_annotation_distance = "0.8" // single character string
of the distance from the center of the slice. 0 means center of the slice, 0.5
means at the edge (positive numeric value ). Above 0.5, the donut will be
reduced to make place for the annotation. Ignored if tree_meta_path is NULL
donut_annotation_size = "4" // single character string of the
annotation font size in mm (numeric value). Ignored if tree_meta_path is NULL
donut_annotation_force = "100" // single character string of
the force of repulsion between overlapping text labels (numeric value). See
?ggrepel::geom_text_repel() in R. Ignored if tree_meta_path is NULL
donut_annotation_force_pull = "1" // single character string
of the force of attraction between a text label and its corresponding data
point (numeric value). See ?ggrepel::geom_text_repel() in R. Ignored if
tree_meta_path is NULL
donut_legend_width = "0.5" // single character string
indicating the relative width of the legend sector (on the right of the plot)
relative to the width of the plot (single proportion between 0 and 1). Value 1
means that the window device width is split in 2, half for the plot and half
for the legend. Value 0 means no room for the legend, which will overlay the
plot region. Write NULL to inactivate the legend sector. In such case, ggplot2
will manage the room required for the legend display, meaning that the width of
the plotting region can vary between graphs, depending on the text in the
legend
donut_legend_text_size = "7" // single character string of the
font size in mm of the legend labels (numeric value)
donut_legend_box_size = "3" // single character string of the
size of the legend squares in mm (numeric value)
donut_legend_box_space = "2" // single character string of the
space between the legend boxes in mm (numeric value)
donut_legend_limit = "0.05" // single character string of the
classes displayed in the legend for which the corresponding proportion is over
the mentionned proportion threshold (positive proportion). Example:
donut_legend_limit = 0.4 means that only the sectors over 40% of the donut will
be in the legend. Write "NULL" for all the sectors in the legend (no
limit required).
}
/*
############################
## ##
##
Local / Cluster ##
## ##
############################
*/
// see
https://confluence.pasteur.fr/pages/viewpage.action?pageId=69304504
system_exec = 'local' // single character
string of the system that runs the workflow. Either 'local' to run on our own
computer or 'slurm' to run on the pasteur cluster. Example: system_exec =
'local'
queue = 'common,dedicated' // single
character string of the -p option of slurm. Example: queue =
'common,dedicated'. Example: queue = 'hubbioit'
qos = '--qos=ultrafast' // single
character string of the --qos option of slurm. Example: qos= '--qos=fast'.
Example: qos = '--qos=ultrafast'. Example: qos = '--qos=hubbioit'
add_options = ' ' // single character
string of the additional option of slurm. Example: add_options =
'--exclude=maestro-1101,maestro-1034' or add_options = ' ', add_options =
'--time=70:00:00' (acceptable time formats include "minutes", "minutes:seconds",
"hours:minutes:seconds", "days-hours",
"days-hours:minutes" and "days-hours:minutes:seconds"). See
https://slurm.schedmd.com/sbatch.html#OPT_time for other options
/*
#############################
## ##
##
Other (optional) ##
## ##
#############################
*/
env{
cute_path =
"https://gitlab.pasteur.fr/gmillot/cute_little_R_functions/-/raw/v12.4.0/cute_little_R_functions.R"
// single character string indicating the file (and absolute pathway) of the
required cute_little_R_functions toolbox. With ethernet connection available,
this can also be used:
"https://gitlab.pasteur.fr/gmillot/cute_little_R_functions/raw/v5.1.0/cute_little_R_functions.R"
or local "C:\\Users\\Gael\\Documents\\Git_projects\\cute_little_R_functions\\cute_little_R_functions.R"
igphylm_exe_path = "/usr/local/share/igphyml/src/igphyml" //
single character string indicating the path of the igphyml exec file. Not it to
change that path when using the containers defined below. Example:
igphylm_exe_path = "/usr/local/share/igphyml/src/igphyml". Example:
igphylm_exe_path =
"\\\\wsl$\\Ubuntu-20.04\\home\\gael\\bin\\igphyml\\src\\igphyml"
}
singularity_path = "NULL" //
single character string of the path of the singularity folder (where all the
singularity images are are pulled and stored for proper nextflow execution).
Write "NULL" for default path (but will not work in most cases).
Example:
singularity_path='/pasteur/zeus/projets/p01/BioIT/gmillot/singularity'.
Example: singularity_path='/mnt/c/Users/gmillot/singularity'. Example:
singularity_path="$baseDir/singularity" # do not forget double quotes
out_path_ini =
"$baseDir/results" // single character string of where the output
files will be saved. Example out_path_ini = '.' for where the main.nf run is
executed or out_path_ini = "$baseDir/results" to put the results in a
result folder (created if required), $baseDir indicating where the main.nf run
is executed. Example: out_path_ini = '/mnt/c/Users/Gael/Desktop'. Example :
out_path_ini="/pasteur/zeus/projets/p01/BioIT/gmillot/08002_bourgeron/results".
Warning: this does not work: out_path_ini = "/mnt/share/Users/gael/Desktop"
result_folder_name="repertoire_profiler"
// single character string.of the name of the folder where the results files
are dorpped
/*
#########################################################################
##
##
##
End Parameters that must be set by the user ##
##
##
#########################################################################
*/
//////// Pre processing
int secs = (new Date().getTime())/1000
out_path="${out_path_ini}/${result_folder_name}_${secs}"
//////// end Pre processing
//////// variables used here and also in
the main.nf file
env {
system_exec = "${system_exec}"
out_path_ini = "${out_path_ini}"
out_path = "${out_path}"
queue = "${queue}"
qos = "${qos}"
add_options = "${add_options}"
}
//////// variables used here and also in
the main.nf file
//////// Scopes
// kind of execution. Either 'local' or
'slurm'
// those are closures. See
https://www.nextflow.io/docs/latest/script.html#closures
executor {
name = "${system_exec}"
queueSize = 2000
}
// create a report folder and print a
html report file . If no absolute path, will be where the run is executed
// see
https://www.nextflow.io/docs/latest/config.html#config-report
report {
enabled = true
file = "${out_path}/reports/report.html" // warning: here
double quotes to get the nextflow variable interpretation
}
// txt file with all the processes and
info
trace {
enabled = true
file = "${out_path}/reports/trace.txt"
}
// html file with all the processes
timeline {
enabled = true
file = "${out_path}/reports/timeline.html"
}
// .dot picture of the workflow
dag {
enabled = true
file = "${out_path}/reports/nf_dag.png"
}
// define singularity parameters
singularity {
enabled = true
autoMounts = true // automatically mounts host paths in the executed
container
if(system_exec == 'slurm' || system_exec == 'slurm_local'){
runOptions = '--no-home --bind /pasteur' //-B /run/shm:/run/shm has been
removed because block the pipeline. Warning: clone_assignment process use
python. Thus, -B /run/shm:/run/shm should be required normally
}else{
runOptions = '--no-home -B /run/shm:/run/shm' // --no-home prevent
singularity to mount the $HOME path and thus forces singularity to work with
only what is inside the container
}
//runOptions = '--home $HOME:/home/$USER --bind /pasteur' // provide any
extra command line options supported by the singularity exec. Here,
fait un bind de tout /pasteur dans /pasteur du container. Sinon pas d acc s
if(singularity_path == "NULL"){
if(system_exec == 'slurm'){
cacheDir =
'/pasteur/zeus/projets/p01/BioIT/gmillot/singularity' // name of the directory
where remote Singularity images are stored. When rerun, the exec directly uses
these without redownloading them. When using a computing cluster it must be a
shared folder accessible to all computing nodes
}else if(system_exec == 'slurm_local'){
cacheDir = 'singularity' //
"$baseDir/singularity" can be used but do not forget double quotes.
}else{
cacheDir =
'/mnt/c/Users/gmillot/singularity' // "$baseDir/singularity" can be
used but do not forget double quotes.
}
}else{
cacheDir = "${singularity_path}"
}
}
//////// end Scopes
//////// directives
// provide the default directives for all
the processes in the main.nf pipeline calling this config file
process {
// directives for all the processes
// executor='local' // no need because already defined above in the
executor scope
if(system_exec == 'slurm'){
queue = "$queue"
clusterOptions = "$qos $add_options"
scratch=false
maxRetries=3
errorStrategy{ task.exitStatus in 137..143 ? 'retry' : 'terminate' }
}else{
maxRetries=0
errorStrategy='terminate'
}
withLabel: bash {
container = 'gmillot/bash-extended_v4.0:gitlab_v8.0'
cpus = 1
memory = '500M'
}
withLabel: immcantation {
container = 'gmillot/immcantation_v1.2:gitlab_v10.1'
cpus = 1
memory = '3G'
}
withLabel: immcantation_10cpu {
container = 'gmillot/immcantation_v1.2:gitlab_v10.1'
cpus = 10
memory = '30G'
}
withLabel: r_ext {
container='gmillot/r_v4.1.2_ig_clustering_v1.3:gitlab_v9.9'
cpus=1 // only used when name = "local" in the executor part
above
memory='3G' // only used when name = "local" in the executor
part above
}
withLabel: seqkit {
container='pegi3s/seqkit:2.3.0'
cpus=1 // only used when name = "local" in the executor part
above
memory='3G' // only used when name = "local" in the executor
part above
}
}
//////// end directives
A way to control number of attempts when failing process, depending on the kind of submission:
process {
queue =
{task.attempt>1 ? "$normalqueue" : "$fastqueue" }
clusterOptions =
{task.attempt > 1 ?
"$normalqos" : "$fastqos" }
time =
{task.attempt>1 ? '10h' : '10m' }
memory={50.GB *
task.attempt}
}